
Control and Prediction of the Organic Solid State
DMACRYS Citations
The information on this page is compiled using the citation report function of Web of Science. It was last updated 19 August 2014. The chart below shows the citations of Price, S. L.; Leslie, M.; Welch, G. W. A.; Habgood, M.; Price, L. S.; Karamertzanis, P. G.; Day, G. M., Modelling organic crystal structures using distributed multipole and polarizability-based model intermolecular potentials. Physical Chemistry Chemical Physics 2010, 12, (30), 8478-8490, by year.
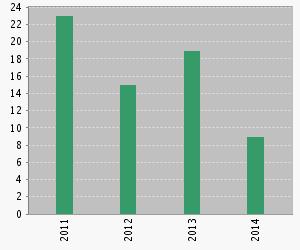
DMAREL Citations
The chart below shows the citations of the original DMAREL paper, Willock, D. J.; Price, S. L.; Leslie, M.; Catlow, C. R. A, The Relaxation of Molecular Crystal Structures Using a Distributed Multipole Electrostatic Model. Journal of Computational Chemistry 1995, 16 (5), 628-647, by year.
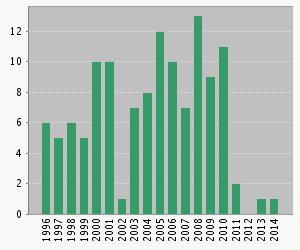
Full citation list
The following articles all cite Price, S. L.; Leslie, M.; Welch, G. W. A.; Habgood, M.; Price, L. S.; Karamertzanis, P. G.; Day, G. M., Modelling organic crystal structures using distributed multipole and polarizability-based model intermolecular potentials. Physical Chemistry Chemical Physics 2010, 12, (30), 8478-8490.
Crystal Structure Prediction
Pharmaceuticals
- Arlin, J. B.; Price, L. S.; Price, S. L.; Florence, A. J., A strategy for producing predicted polymorphs: catemeric carbamazepine form V. Chemical Communications 2011, 47, (25), 7074-7076.
- Baias, M.; Dumez, J. N.; Svensson, P. H.; Schantz, S.; Day, G. M.; Emsley, L., De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography. Journal of the American Chemical Society 2013, 135, (46), 17501-17507.
- Baias, M.; Widdifield, C. M.; Dumez, J. N.; Thompson, H. P. G.; Cooper, T. G.; Salager, E.; Bassil, S.; Stein, R. S.; Lesage, A.; Day, G. M.; Emsley, L., Powder crystallography of pharmaceutical materials by combined crystal structure prediction and solid-state H-1 NMR spectroscopy. Physical Chemistry Chemical Physics 2013, 15, (21), 8069-8080.
- Bhardwaj, R. M.; Price, L. S.; Price, S. L.; Reutzel-Edens, S. M.; Miller, G. J.; Oswald, I. D. H.; Johnston, B. F.; Florence, A. J., Exploring the Experimental and Computed Crystal Energy Landscape of Olanzapine. Crystal Growth & Design 2013, 13, (4), 1602-1617.
- Braun, D. E.; Ardid-Candel, M.; D'Oria, E.; Karamertzanis, P. G.; Arlin, J. B.; Florence, A. J.; Jones, A. G.; Price, S. L., Racemic Naproxen: A Multidisciplinary Structural and Thermodynamic Comparison with the Enantiopure Form. Crystal Growth & Design 2011, 11, (12), 5659-5669.
- Braun, D. E.; McMahon, J. A.; Koztecki, L. H.; Price, S. L.; Reutzel-Edens, S. M., Contrasting Polymorphism of Related Small Molecule Drugs Correlated and Guided by the Computed Crystal Energy Landscape. Crystal Growth & Design 2014, 14, (4), 2056-2072.
- Eddleston, M. D.; Patel, B.; Day, G. M.; Jones, W., Cocrystallization by Freeze-Drying: Preparation of Novel Multicomponent Crystal Forms. Crystal Growth & Design 2013, 13, (10), 4599-4606.
- Eddleston, M. D.; Hejczyk, K. E.; Bithell, E. G.; Day, G. M.; Jones, W., Determination of the Crystal Structure of a New Polymorph of Theophylline. Chemistry-a European Journal 2013, 19, (24), 7883-7888.
- Eddleston, M. D.; Hejczyk, K. E.; Bithell, E. G.; Day, G. M.; Jones, W., Polymorph Identification and Crystal Structure Determination by a Combined Crystal Structure Prediction and Transmission Electron Microscopy Approach. Chemistry-a European Journal 2013, 19, (24), 7874-7882.
- Habgood, M., Analysis of Enantiospecific and Diastereomeric Cocrystal Systems by Crystal Structure Prediction. Crystal Growth & Design 2013, 13, (10), 4549-4558.
- Ismail, S. Z.; Anderton, C. L.; Copley, R. C. B.; Price, L. S.; Price, S. L., Evaluating a Crystal Energy Landscape in the Context of Industrial Polymorph Screening. Crystal Growth & Design 2013, 13, (6), 2396-2406.
- Johnston, A.; Bardin, J.; Johnston, B. F.; Fernandes, P.; Kennedy, A. R.; Price, S. L.; Florence, A. J., Experimental and Predicted Crystal Energy Landscapes of Chlorothiazide. Crystal Growth & Design 2011, 11, (2), 405-413.
- Uzoh, O. G.; Cruz-Cabeza, A. J.; Price, S. L., Is the Fenamate Group a Polymorphophore? Contrasting the Crystal Energy Landscapes of Fenamic and Tolfenamic Acids. Crystal Growth & Design 2012, 12, (8), 4230-4239.
- Wu, H.; Habgood, M.; Parker, J. E.; Reeves-McLaren, N.; Cockcroft, J. K.; Vickers, M.; West, A. R.; Jones, A. G., Crystal structure determination by combined synchrotron powder X-ray diffraction and crystal structure prediction: 1: 1 L-ephedrine D-tartrate. Crystengcomm 2013, 15, (10), 1853-1859.
Small molecules
- Braun, D. E.; Karamertzanis, P. G.; Arlin, J. B.; Florence, A. J.; Kahlenberg, V.; Tocher, D. A.; Griesser, U. J.; Price, S. L., Solid-State Forms of beta-Resorcylic Acid: How Exhaustive Should a Polymorph Screen Be? Crystal Growth & Design 2011, 11, (1), 210-220.
- Braun, D. E.; Karamertzanis, P. G.; Price, S. L., Which, if any, hydrates will crystallise? Predicting hydrate formation of two dihydroxybenzoic acids. Chemical Communications 2011, 47, (19), 5443-5445.
- Braun, D. E.; Tocher, D. A.; Price, S. L.; Griesser, U. J., The Complexity of Hydration of Phloroglucinol: A Comprehensive Structural and Thermodynamic Characterization. Journal of Physical Chemistry B 2012, 116, (13), 3961-3972.
- Braun, D. E.; Bhardwaj, R. M.; Arlin, J. B.; Florence, A. J.; Kahlenberg, V.; Griesser, U. J.; Tocher, D. A.; Price, S. L., Absorbing a Little Water: The Structural, Thermodynamic, and Kinetic Relationship between Pyrogallol and Its Tetarto-Hydrate. Crystal Growth & Design 2013, 13, (9), 4071-4083.
- Braun, D. E.; Bhardwaj, R. M.; Florence, A. J.; Tocher, D. A.; Price, S. L., Complex Polymorphic System of Gallic Acid-Five Monohydrates, Three Anhydrates, and over 20 Solvates. Crystal Growth & Design 2013, 13, (1), 19-23.
- Cruz-Cabeza, A. J.; Schwalbe, C. H., Observed and predicted hydrogen bond motifs in crystal structures of hydantoins, dihydrouracils and uracils. New Journal of Chemistry 2012, 36, (6), 1347-1354.
- Cruz-Cabeza, A. J.; Liebeschuetz, J. W.; Allen, F. H., Systematic conformational bias in small-molecule crystal structures is rare and explicable. Crystengcomm 2012, 14, (20), 6797-6811.
- Habgood, M.; Lancaster, R. W.; Gateshki, M.; Kenwright, A. M., The Amorphous Form of Salicylsalicylic Acid: Experimental Characterization and Computational Predictability. Crystal Growth & Design 2013, 13, (4), 1771-1779.
- Issa, N.; Barnett, S. A.; Mohamed, S.; Braun, D. E.; Copley, R. C. B.; Tocher, D. A.; Price, S. L., Screening for cocrystals of succinic acid and 4-aminobenzoic acid. Crystengcomm 2012, 14, (7), 2454-2464.
- Mohamed, S.; Tocher, D. A.; Price, S. L., Computational prediction of salt and cocrystal structures-Does a proton position matter? International Journal of Pharmaceutics 2011, 418, (2), 187-198.
- Ridout, J.; Price, L. S.; Howard, J. A. K.; Probert, M. R., Polymorphism Arising from Differing Rates of Compression of Liquids. Crystal Growth & Design 2014, 14, (7), 3384-3391.
- Spencer, J.; Patel, H.; Deadman, J. J.; Palmer, R. A.; Male, L.; Coles, S. J.; Uzoh, O. G.; Price, S. L., The unexpected but predictable tetrazole packing in flexible 1-benzyl-1H-tetrazole. Crystengcomm 2012, 14, (20), 6441-6446.
- Vasileiadis, M.; Kazantsev, A. V.; Karamertzanis, P. G.; Adjiman, C. S.; Pantelides, C. C., The polymorphs of ROY: application of a systematic crystal structure prediction technique. Acta Crystallographica Section B-Structural Science 2012, 68, 677-685.
Porous organic cages
- Jones, J. T. A.; Hasell, T.; Wu, X. F.; Bacsa, J.; Jelfs, K. E.; Schmidtmann, M.; Chong, S. Y.; Adams, D. J.; Trewin, A.; Schiffman, F.; Cora, F.; Slater, B.; Steiner, A.; Day, G. M.; Cooper, A. I., Modular and predictable assembly of porous organic molecular crystals. Nature 2011, 474, (7351), 367-371.
- Pyzer-Knapp, E. O.; Thompson, H. P. G.; Schiffmann, F.; Jelfs, K. E.; Chong, S. Y.; Little, M. A.; Cooper, A. I.; Day, G. M., Predicted crystal energy landscapes of porous organic cages. Chemical Science 2014, 5, (6), 2235-2245.
Mixed work, including blind tests
- Bardwell, D. A.; Adjiman, C. S.; Arnautova, Y. A.; Bartashevich, E.; Boerrigter, S. X. M.; Braun, D. E.; Cruz-Cabeza, A. J.; Day, G. M.; Della Valle, R. G.; Desiraju, G. R.; van Eijck, B. P.; Facelli, J. C.; Ferraro, M. B.; Grillo, D.; Habgood, M.; Hofmann, D. W. M.; Hofmann, F.; Jose, K. V. J.; Karamertzanis, P. G.; Kazantsev, A. V.; Kendrick, J.; Kuleshova, L. N.; Leusen, F. J. J.; Maleev, A. V.; Misquitta, A. J.; Mohamed, S.; Needs, R. J.; Neumann, M. A.; Nikylov, D.; Orendt, A. M.; Pal, R.; Pantelides, C. C.; Pickard, C. J.; Price, L. S.; Price, S. L.; Scheraga, H. A.; van de Streek, J.; Thakur, T. S.; Tiwari, S.; Venuti, E.; Zhitkov, I. K., Towards crystal structure prediction of complex organic compounds - a report on the fifth blind test. Acta Crystallographica Section B-Structural Science 2011, 67, 535-551.
- Kazantsev, A. V.; Karamertzanis, P. G.; Adjiman, C. S.; Pantelides, C. C.; Price, S. L.; Galek, P. T. A.; Day, G. M.; Cruz-Cabeza, A. J., Successful prediction of a model pharmaceutical in the fifth blind test of crystal structure prediction. International Journal of Pharmaceutics 2011, 418, (2), 168-178.
Energy evaluation
- Belenguer, A. M.; Friscic, T.; Day, G. M.; Sanders, J. K. M., Solid-state dynamic combinatorial chemistry: reversibility and thermodynamic product selection in covalent mechanosynthesis. Chemical Science 2011, 2, (4), 696-700.
- Galcera, J.; Friscic, T.; Hejczyk, K. E.; Fabian, L.; Clarke, S. M.; Day, G. M.; Molins, E.; Jones, W., Isostructural organic binary-host frameworks with tuneable and diversely decorated inclusion cavities. Crystengcomm 2012, 14, (23), 7898-7906.
- Gelbrich, T.; Braun, D. E.; Ellern, A.; Griesser, U. J., Four Polymorphs of Methyl Paraben: Structural Relationships and Relative Energy Differences. Crystal Growth & Design 2013, 13, (3), 1206-1217.
Development of method in other workflows
- Habgood, M., Form II Caffeine: A Case Study for Confirming and Predicting Disorder in Organic Crystals. Crystal Growth & Design 2011, 11, (8), 3600-3608.
- Habgood, M., Solution and nanoscale structure selection: implications for the crystal energy landscape of tetrolic acid. Physical Chemistry Chemical Physics 2012, 14, (25), 9195-9203.
- Habgood, M.; Grau-Crespo, R.; Price, S. L., Substitutional and orientational disorder in organic crystals: a symmetry-adapted ensemble model. Physical Chemistry Chemical Physics 2011, 13, (20), 9590-9600.
- Habgood, M.; Price, S. L.; Portalone, G.; Irrera, S., Testing a Variety of Electronic-Structure-Based Methods for the Relative Energies of 5-Formyluracil Crystals. Journal of Chemical Theory and Computation 2011, 7, (9), 2685-2688.
- Habgood, M., Intermolecular electrostatics via molecular fragmentation: an application to crystal structure prediction. Molecular Simulation 2012, 38, (13), 1103-1113.
- Kazantsev, A. V.; Karamertzanis, P. G.; Adjiman, C. S.; Pantelides, C. C., Efficient Handling of Molecular Flexibility in Lattice Energy Minimization of Organic Crystals. Journal of Chemical Theory and Computation 2011, 7, (6), 1998-2016.
- McDonagh, J. L.; Nath, N.; De Ferrari, L.; van Mourik, T.; Mitchell, J. B. O., Uniting Cheminformatics and Chemical Theory To Predict the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules. Journal of Chemical Information and Modeling 2014, 54, (3), 844-856
- Palmer, D. S.; McDonagh, J. L.; Mitchell, J. B. O.; van Mourik, T.; Fedorov, M. V., First-Principles Calculation of the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules. Journal of Chemical Theory and Computation 2012, 8, (9), 3322-3337.
- Zhu, Q.; Oganov, A. R.; Glass, C. W.; Stokes, H. T., Constrained evolutionary algorithm for structure prediction of molecular crystals: methodology and applications. Acta Crystallographica Section B-Structural Science 2012, 68, 215-226.
Review articles and Book chapters
- Abramov, Y. A., Current Computational Approaches to Support Pharmaceutical Solid Form Selection. Organic Process Research & Development 2013, 17, (3), 472-485.
- Brittain, H. G., Polymorphism and solvatomorphism 2010. Journal of Pharmaceutical Sciences 2012, 101, (2), 464-484.
- Cisneros, G. A.; Karttunen, M.; Ren, P. Y.; Sagui, C., Classical Electrostatics for Biomolecular Simulations. Chemical Reviews 2014, 114, (1), 779-814.
- Day, G. M., Current approaches to predicting molecular organic crystal structures. Crystallography Reviews 2011, 17, (1), 3-52.
- Leusen, F. J. J.; Kendrick, J., Polymorph Prediction of Small Organic Molecules, Co-crystals and Salts. In Pharmaceutical Salts and Co-Crystals, Wouters, J.; Quere, L., Eds. 2011; pp 44-88.
- Mitchell, J. B., Informatics, machine learning and computational medicinal chemistry. Future Medicinal Chemistry 2011, 3, (4), 451-467.
- Price, S. L.; Price, L. S., Computational Polymorph Prediction. In Solid State Characterisation of Pharmaceuticals, Storey, R.; Ymen, I., Eds. 2011; p 427-450.
- Price, S. L., Why don't we find more polymorphs? Acta Crystallographica Section B-Structural Science 2013, 69, 313-328.
- Price, S. L., Predicting crystal structures of organic compounds. Chemical Society Reviews 2014, 43, (7), 2098-2111.
Papers that cite, but don't use DMACRYS
- Cardamone, S.; Hughes, T. J.; Popelier, P. L. A., Multipolar electrostatics. Physical Chemistry Chemical Physics 2014, 16, (22), 10367-10387.
- Kandathil, S. M.; Fletcher, T. L.; Yuan, Y. N.; Knowles, J.; Popelier, P. L. A., Accuracy and Tractability of a Kriging Model of Intramolecular Polarizable Multipolar Electrostatics and Its Application to Histidine. Journal of Computational Chemistry 2013, 34, (21), 1850-1861.
- Kramer, C.; Gedeck, P.; Meuwly, M., Atomic multipoles: Electrostatic potential fit, local reference axis systems, and conformational dependence. Journal of Computational Chemistry 2012, 33, (20), 1673-1688.
- Kramer, C.; Bereau, T.; Spinn, A.; Liedl, K. R.; Gedeck, P.; Meuwly, M., Deriving Static Atomic Multipoles from the Electrostatic Potential. Journal of Chemical Information and Modeling 2013, 53, (12), 3410-3417.
- Kramer, C.; Gedeck, P.; Meuwly, M., Multipole-Based Force Fields from ab Initio Interaction Energies and the Need for Jointly Refitting All Intermolecular Parameters. Journal of Chemical Theory and Computation 2013, 9, (3), 1499-1511.
- Le, H. A.; Bettens, R. P. A., Distributed Multipoles and Energies of Flexible Molecules. Journal of Chemical Theory and Computation 2011, 7, (4), 921-930. - incorrect citation?
- Mocilac, P.; Gallagher, J. F., Structural systematics and conformational analyses of a 3 x 3 isomer grid of nine N-(tolyl)pyridinecarboxamides and three chlorinated relatives. Crystengcomm 2011, 13, (17), 5354-5366.
- Ruhle, V.; Kusumaatmaja, H.; Chakrabarti, D.; Wales, D. J., Exploring Energy Landscapes: Metrics, Pathways, and Normal-Mode Analysis for Rigid-Body Molecules. Journal of Chemical Theory and Computation 2013, 9, (9), 4026-4034.
- Ryno, S. M.; Risko, C.; Bredas, J. L., Impact of Molecular Packing on Electronic Polarization in Organic Crystals: The Case of Pentacene vs TIPS-Pentacene. Journal of the American Chemical Society 2014, 136, (17), 6421-6427.
- Tafipolsky, M.; Engels, B., Accurate Intermolecular Potentials with Physically Grounded Electrostatics. Journal of Chemical Theory and Computation 2011, 7, (6), 1791-1803.
- Totton, T. S.; Misquitta, A. J.; Kraft, M., Assessing the Polycyclic Aromatic Hydrocarbon Anisotropic Potential with Application to the Exfoliation Energy of Graphite. Journal of Physical Chemistry A 2011, 115, (46), 13684-13693.
- Wen, S. H.; Beran, G. J. O., Accurate Molecular Crystal Lattice Energies from a Fragment QM/MM Approach with On-the-Fly Ab Initio Force Field Parametrization. Journal of Chemical Theory and Computation 2011, 7, (11), 3733-3742.
- Wen, S. H.; Nanda, K.; Huang, Y. H.; Beran, G. J. O., Practical quantum mechanics-based fragment methods for predicting molecular crystal properties. Physical Chemistry Chemical Physics 2012, 14, (21), 7578-7590.
- Yuan, Y. N.; Mills, M. J. L.; La Popelier, P., Multipolar electrostatics based on the Kriging machine learning method: an application to serine. Journal of Molecular Modeling 2014, 20, (4).
Enter secure pages (For project members only - password required)
© UCL Chemistry Department 2004. This page was last updated on 17 August, 2017. If you have any problems with this page please email the WebMaster