
Control and Prediction of the Organic Solid State
Finding polymorphs using computational Crystal Structure Prediction
If we use Crystal Structure Prediction (CSP) methods and predict a polymorph should exist, how do we nucleate it? The EPSRC project "Computationally Designed Templates for Exquisite Control of Polymorphic Form" was inspired by the success of the CPOSS project at targeting the first polymorph of carbamazepine which does not have hydrogen-bonded dimers. It seeks to establish how, given a predicted polymorph, we can design the experiment to find it. It continues to develop the capability of calculating and interpreting Crystal Energy Landscapes, the set of thermodynamically plausible structures generated by CSP, with increasing accuracy for a range of organic systems.
The MagnaPharm project is looking at the effect of magnetic field on crystal nucleation as a potential route to predicted polymorphs.
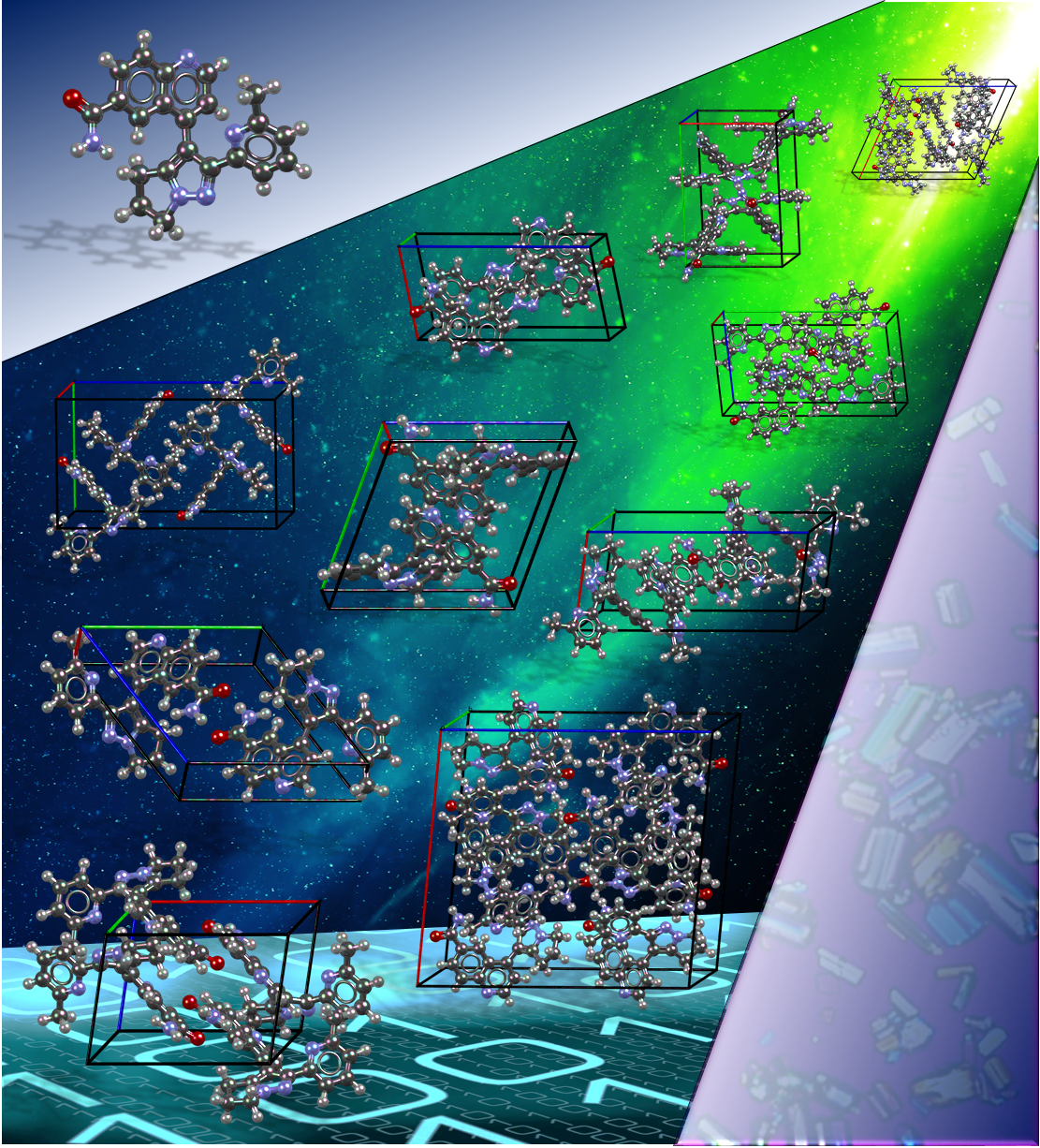
Crystal Energy Landscapes can confirm that the most thermodynamically stable form is known, be used to help solve crystal structures from powder X-ray diffraction, suggest the occurrence of disorder in the crystal lattice, as well as suggesting which other polymorphs might be found. We are working to establish the effective use of these landscapes as a complement to academic and industrial solid form screening. This site contains information on the calculation of crystal energy landscapes including the programmes used, our database of calculated crystal structures and publications.
We thank for EPSRC Basic Technology Program and the CPOSS Industrial Alliance for funding "Control and Prediction of the Organic Solid State" from October 2003 to September 2012. The project is closely aligned with the EPSRC Directed Assembly Network (theme 2) of "Beyond the Molecule", which organises meetings and provides funding for collaborations, and with the EPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation to facilitate Knowledge Transfer to industry.
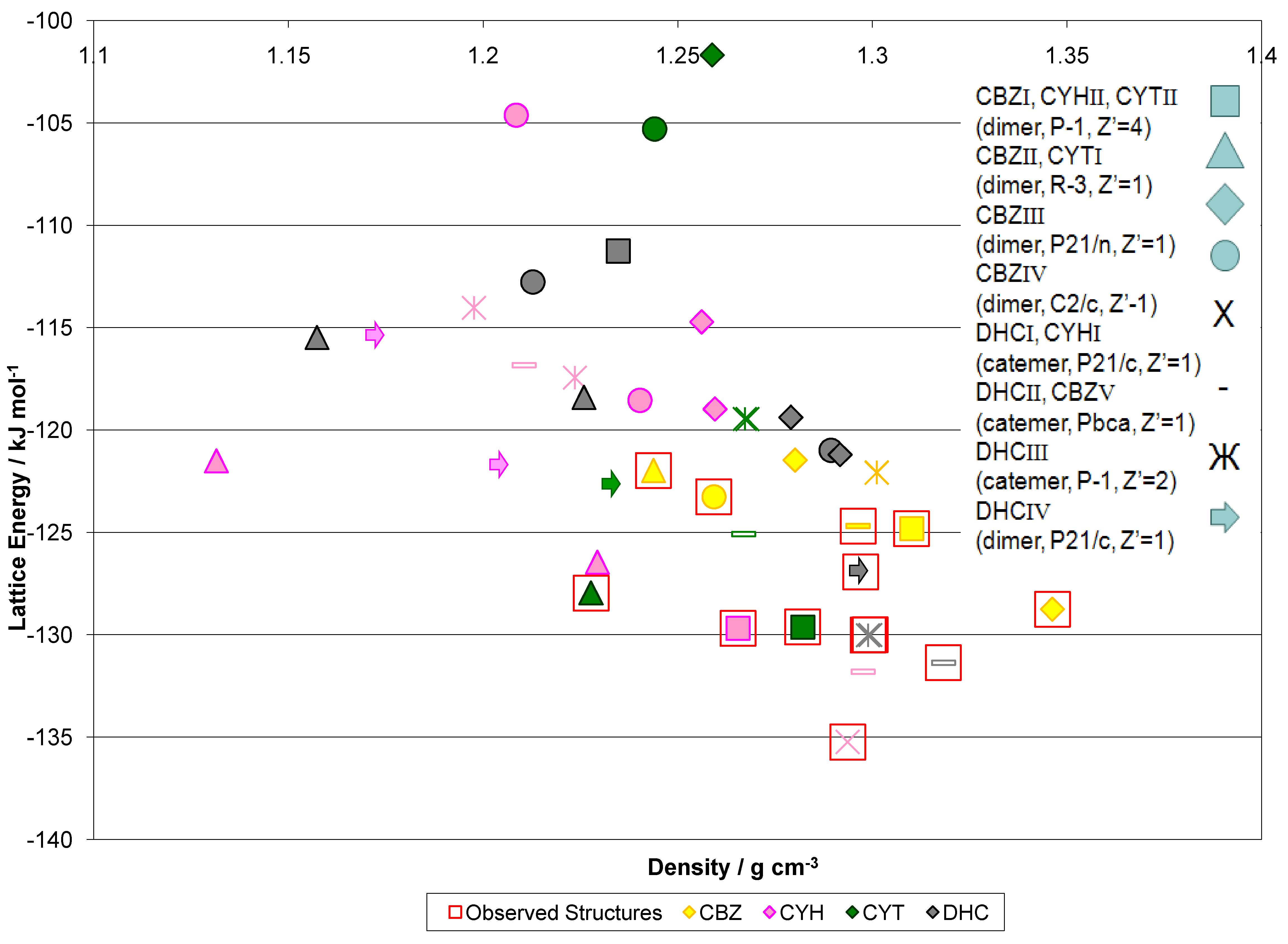
We have been working closely with scientists at Eli-Lilly for some time now, and a recent piece of work was published in The Journal of the American Chemical Society.
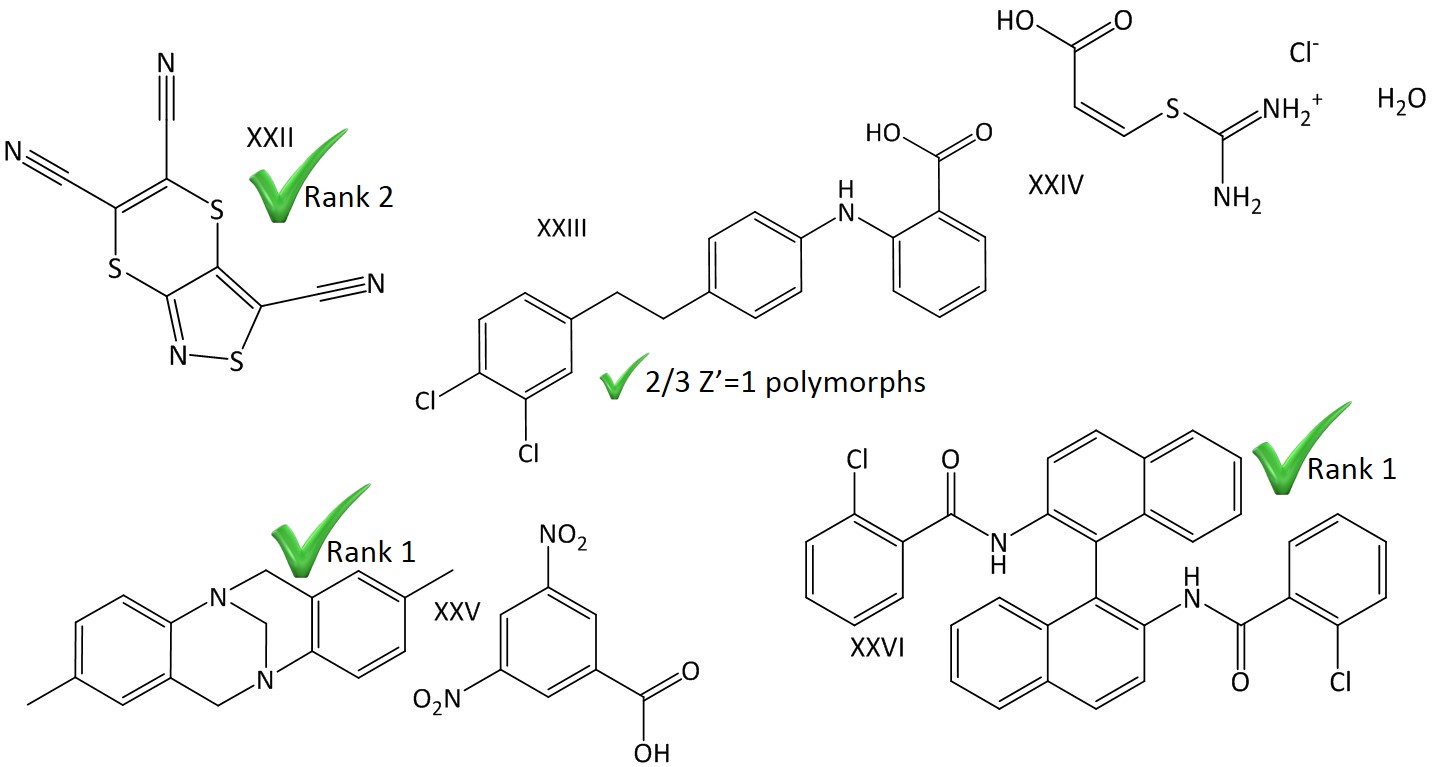
| |||||||
The project was well-represented at the recent Faraday Discussion on Crystal Structure Prediction, including the introductory lecture and an invited contribution.
| |||||||
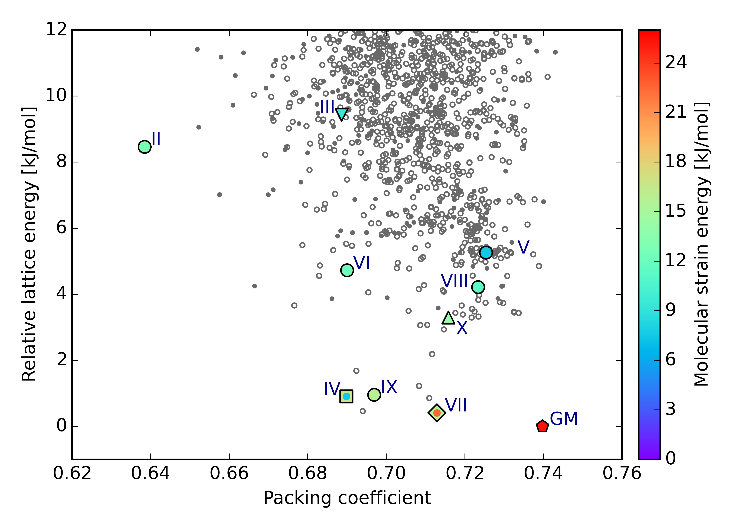
 | We have shown that we can target predicted polymorphs, such as form III of cyheptamide, by sublimation onto an isomorphous template. See: Isomorphous template induced crystallisation: a robust method for the targeted crystallisation of computationally predicted metastable polymorphs (Srirambhatla, V. K.; Guo, R.; Price, S. L.; Florence, A. J., Chemical Communications 2016, 52, 7384-7386) |
|
|||||
| We continue to develop methods of Crystal Structure Prediction, and are pleased by our success in the 6th Blind Test. We are continuing to extend our ability to model more complex systems, to calculate relative thermodynamic stability with the latest advances in computational chemistry, and to use CSP methods to investigate crystallization. The article "Report on the sixth blind test of organic crystal structure prediction methods" has now been published as Reilly, A. M. et al, Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials, 2016, Volume B72, pages 439-459 | ||||||
Enter secure pages (For project members only - password required)
© UCL Chemistry Department 2004. This page was last updated on 4 September, 2019. If you have any problems with this page please email the WebMaster