Emerging Classifications of Polymorph Predictability
"Computed
crystal energy landscapes for understanding and predicting organic crystal
structures and polymorphism." Price SL 2009. Accounts Chem Res 42:117-126.
summarises the evidence for the interpretation of energy landscapes.
Each computational study starts by generating
a set of energetically feasible crystal structures for the molecule.
We are currently limited in the molecules
that can be studied, and the types
of crystal structure that can be found in the search, as well as the
accuracy of the relative energies.
The resulting distributions of the lattice energies of the known and hypothetical
crystal structures fall into different (overlapping) categories, which
differ in the confidence with which the crystal structures can be predicted.
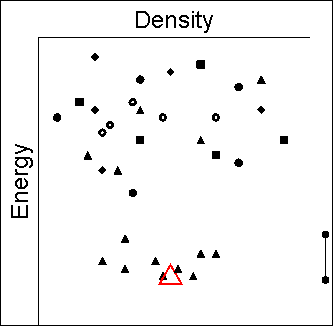
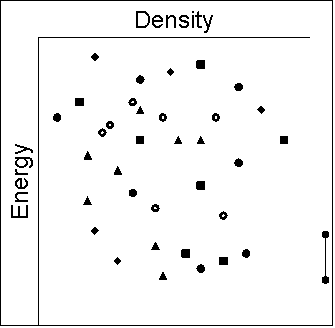
Let us assume that the following schematic
diagrams give the relative energies
for all different crystal structures, with open red shapes denoting the
experimentally observed structures and shape of symbols depicting groups
of similar structures. Such diagrams summarise the crystal energy landscape,
the structures and relative energies of the thermodynamically plausible
crystal packings of the molecule. The bar to the right of each plot indicates
the energy range of potential polymorphism.
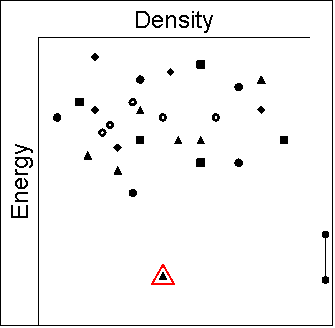 |
- The only known crystal structure is found at the global
minimum and there is a sufficient energy gap that no others
are energetically plausible. Only in this case is the search
for energetically feasible structures able to unambiguously
predict the structure and rule out the possibility of polymorphism.
Examples below show the gap to the next hypothetical structure
found in the search:
- 3-oxauracil (4 kJ mol-1)
- 1,2-dichloro-3-nitrobenzene (1.7 kJ mol-1)
|
|
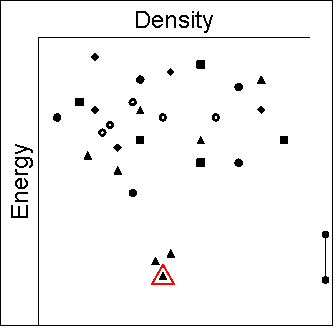
- There is a known crystal structure at the global minimum, and
there are other structures close enough in energy that they could
be potential polymorphs. Either:
- the other low energy crystal structures are closely related,
and so it is unlikely that the nucleation pathway would
produce a metastable structure, and even if it did it would
be likely to transform to the more stable structure fairly
easily. In this case polymorphism is unlikely.
- Imidazole (same hydrogen
bonded chain)
- 5-azauracil (same hydrogen bonded sheet)
- 5-hydroxyuracil
- Coumarin
- 1,3-dichloro-5-nitrobenzene
- 7-hydroxycoumarin
|
 |
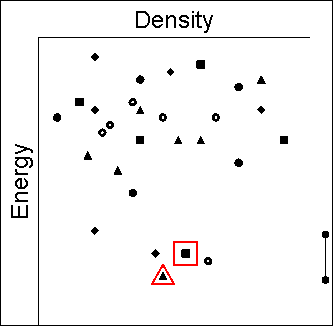
- some of the other hypothetical structures are sufficiently
different (e.g. in hydrogen bonding motif) that it is plausible
that different crystallisation conditions might nucleate
different structures and that transforming to the most stable
structure would be difficult. Hence, we need to rationalise
the kinetic factors that can lead to the observation of
metastable polymorphs. This may be linked to the range of
crystallisation conditions that can be used.
- Polymorphic
- No polymorphs observed so far, but very limited range
of crystallisation conditions available.
- uracil (no polymorphs observed
so far, but limited solubility)
- 6-azauracil (no polymorphs
observed so far, but limited solubility)
- alloxan (no polymorphs observed so far, but reacts
readily)
- parabanic acid
- Some other rationalisation for kinetic effects preventing
polymorphism
- 3-azabicyclononane-2,4-dione
- 5-fluoroisatin
|
 |
|
- The known crystal structure is not the global minimum, i.e.
there are other structures that are predicted to be more stable.
This would be a valuable warning that there is potentially a more
stable polymorph, and by having the predicted structure it may
be possible to design methods of crystallisation to find the new
polymorph.
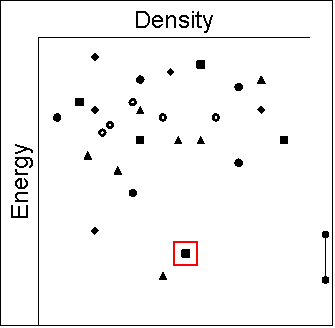 |
- Cases where new polymorphs have been found following the
prediction:
- Cases where no new polymorphs have yet been found, and
we need to understand why there appears to be no kinetic
pathway to the most thermodynamically stable form. This
again may reflect the variety of crystallisation conditions,
for example
- 2,6-diamino-3,5-dinitropyrazine (alternative hydrogen
bonding patterns observed)
- cyanuric acid (sheets preferred)
- p-dichlorobenzene (growth rate favours known structures)
- p-hydroxyuracil (alternative hydrogen bonding patterns)
- adenine (poorly soluble)
- guanine (poorly soluble)
|
|
| Note that the cases 2b and 3 differ
in whether it is easy to crystallise the most thermodynamically
stable polymorph. In both cases, we need extensive experimental
studies seeking to identify all long-lived polymorphs in order to
understand the kinetic factors involved. Following extensive experimental
studies (even those where reactions have been carried out with no
new polymorphs being identified), it should be possible to develop
a more quantitative and predictive computational model of the kinetic
factors that determine polymorphism. This is a major aim of the
CPOSS project. |
- There are a multitude of hypothetical structures very close
together near the energy minimum.
- The experimental structure is not the sort of simple structure
that can be found in the theoretical search. However, there
is a relationship between the hypothetical low energy structures
that enables packing motifs to interchange, e.g. different
stacking of sheets. In this case the predictions suggest
that a specific type of disorder is likely in the crystal.
For example:
- cyclopentane (rotational disorder)
- chlorothalonil (Form 2: disordered sheets; Form 3:
Z'=3 - both closely related to pairs of structures found
in the search)
- 5-chlorouracil
- 5-bromouracil
- azetidine (the Z'=2 structure is probably a thermal
average over Z'=4 lower symmetry structures)
- aspirin (evidence of polymorphic domains of the 2
forms has been found)
- carbonic acid (with 2 distinct amorphous phases linked
to the two polymorphs of unknown structure)
|
 |
- The poor lattice energy and multitude of equally poor
compromises between close packing and the specific interactions
show that the molecule has no good ways of packing with
itself. The hydrogen bonding motifs seen in these low energy
structures are observed in a range of polymorphs and solvates.
- hydrochlorothiazide
- 5-fluorocytosine
|
 |
|
There are cases where we had hoped to be able to predict the
crystal structure, but it became obvious during the course of the
calculations that the accuracy in the relative energies was such
that the assignment into one of the above categories would be very
tentative. Possible reasons include:
- the relative energies of the low energy crystal structures are
very sensitive to small
changes in the molecular conformation, which may be induced by
the packing forces, for example:
- barbituric acid
- 5-hydroxyuracil
- hydantoin
- uric acid
- urazole
In many of these cases the observed crystal structure is the global
minimum structure when the experimental molecular conformation
is used, but is significantly above the global minimum observed
using the gas phase conformer.
- the intermolecular potential is not known sufficiently accurately
so that different reasonable models produce qualitatively different
energy diagrams, for example:
- parabanic acid
- 2,9-di-iodo-anthanthrone (tackled for the Blind Test)
- The calculation of the free energy, rather than the lattice
energy, produces a significant reordering of the structures.
|
We are currently developing our method of crystal structure prediction
and methods of modelling both intermolecular and intramolecular forces,
to both model the above systems better, and progress to salts, hydrates,
conformationally flexible molecules, and more complicated systems and
more diverse molecules. The experimental studies being performed on such
systems are essential to the development of predictive methods, as many
screening studies are finding new forms.
|




