




Repulsion Dispersion Potentials
Most of the crystal structure modelling using DMAREL & DMACRYS that has not derived a specific potential for the molecule1,2 has supplemented the Distributed Multipole electrostatic model with an empirically fitted isotropic atom-atom potential of the form:
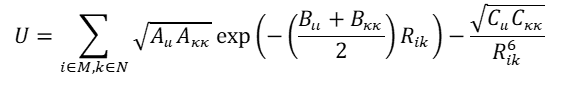
where atom i of type i in molecule M is separated by Rik from atom k of type k in molecule N. Some notes and comments on their use follow, with references to the definitive literature.
WILL01 was derived3 by fitting to a wide range of crystal structures and validated against peptide and nucleoside structures. A key feature is that the hydrogen interaction sites are moved by 0.1 Å into the H-X bond from their neutron or ab initio optimised positions. DMACRYS can perform this foreshortening, which has to also be used in the DMA analysis.
| Potential |
Atom pair |
Description |
Aik /kJ mol-1 |
Bik/Å-1 |
Cik/kJ mol-1 Å6 |
| WILL01 |
C(2)···C(2) |
C bonded to 2 atoms |
103235 |
3.60 |
1435.09 |
| WILL01 |
C(3)···C(3) |
C bonded to 3 atoms |
270363 |
3.60 |
1701.73 |
| WILL01 |
C(4)···C(4) |
C bonded to 4 atoms |
131571 |
3.60 |
978.36 |
| WILL01 |
H(1)···H(1) |
H bonded to C |
12680 |
3.56 |
278.37 |
| WILL01 |
H(2)···H(2) |
H in alcoholic group |
361.30 |
3.56 |
0 |
| WILL01 |
H(3)···H(3) |
H in carboxyl group |
115.70 |
3.56 |
0 |
| WILL01 |
H(4)···H(4) |
H in N-H group |
764.90 |
3.56 |
0 |
| WILL01 |
N(1)···N(1) |
N in triple bond |
96349 |
3.48 |
1407.57 |
| WILL01 |
N(2)···N(2) |
other N with no bonded H |
102369 |
3.48 |
1398.15 |
| WILL01 |
N(3)···N(3) |
N bonded to 1 H atom |
191935 |
3.48 |
2376.55 |
| WILL01 |
N(4)···N(4) |
N with 2 bonded H atoms |
405341 |
3.48 |
5629.82 |
| WILL01 |
O(1)···O(1) |
O bonded to 1 other atom |
241042 |
3.96 |
1260.73 |
| WILL01 |
O(2)···O(2) |
O bonded to 2 other atoms |
284623 |
3.96 |
1285.87 |
This potential was successfully used in a large survey of crystal structure predictions in conjunction with both a point charge4 and Distributed Multipole model5. However problems have been observed in underestimating hydrogen bond distances when combined with a Distributed Multipole Model, presumably because the electrostatic forces can be stronger than with the point charge model used in Williams' parameterisation. In certain cases, of (carboxylate)O···H-O and N···H-O hydrogen bonds the underestimate has led to the hydrogen bonds becoming within the covalent bond range. (An ad hoc fix for the latter6 is to replace the purely repulsive, Williams potential between pyridine nitrogen and carboxylic proton (N(2).H(3)) to A=75719.47 kJ/mol and B=5.1765 Å-1 to model the cocrystal of 4-aminobenzoic acid with 4-nitrophenylacetic acid. This potential is much steeper at unphysically short distances without penalising the lattice energy at typical contacts compared with the original Williams parameterisation.) Graeme Day's group noted noted that O-H(3)···N(2) distances were unreasonably short for the cocrystal in the 2007 blindtest, and found substituting the alcohol H(2) parameters for H(3) in this carboxylic acid produced more reasonable results. We are currently testing the published WILL01 potentials for a range of systems, to establish whether it is worth providing a revision of all the polar hydrogen parameters for use in conjunction with a DMA.
FIT has evolved through using Williams older parameterisations, which had each element in conjunction with C and H only. The H nuclei are used as the interaction sites, although whether Williams corrected for the X-ray foreshortening in these derivations is unclear.
| Reference |
Atom pair |
Description |
Aik /kJ mol-1 |
Bik/Å-1 |
Cik/kJ mol-1 Å6 |
| 7 |
C···C |
Any C atom |
369743 |
3.60 |
2439.8 |
| 7 |
H···H |
H bonded to C |
11971 |
3.74 |
136.4 |
| 8 |
HN···HN |
H bonded to N |
5029.68 |
4.66 |
21.50 |
| 9 |
HO···HO |
H bonded to O |
2263.3 |
4.66 |
21.50 |
| 7 |
N···N |
Any N atom |
254529 |
3.78 |
1378.4 |
| 10 |
O···O |
Any O atom |
230064.1 |
c=3.96 |
1123.59 |
| 11 |
F···F |
Any F atom |
363725 |
4.16 |
844 |
| 12 |
Cl···Cl |
Any Cl Atom |
924675 |
c=3.51 |
7740.48 |
| Other parameters that have been used in conjunction with FIT for a few applications |
|||||
| 16 |
S···S |
S atoms in blind tests (for thiol and thioether) |
401033.7336 |
3.30 |
5790.656 |
| 17 |
S···S |
S atoms in sulfoxides |
236501.5221 |
2.90 |
8397.1147 |
| 17 |
O···O |
O atoms in sulfoxides |
202983.6471 |
3.96 |
640.8628309 |
Note that NEIGHBOURS does not distinguish between H-O and H-N and so many papers have use the HN (=Hp) parameters for any polar hydrogen, including in hydrates and ice.13 NEIGHCRYS will automatically provide the Williams typing and can allow a user specified typing, allowing the ability of DMACRYS to use more atomic types for a given atom to be applied without manual editing of the input file. The extent to which the parameters in combination have been tested beyond8 is limited, though there has been some validation for the F parameters in Ashley Hulme's thesis and for the isotropic chlorine in situations without close Cl···Cl contacts.14 There are various failures: problems in stacking of some rigid aromatics differences led Tom Lewis to reduce the C parameters by 25% in his studies (thesis + 15 ).
It is important to note that both these empirically fitted potentials are effectively modelling the total intermolecular potential with the electrostatic component removed, as well as it is sampled in the crystal structures used for fitting and validation. It is not surprising that the results can be sensitive to the quality of the electrostatic model used, and may be very poor for atypical short contacts. Since they are empirically based, the choice of which to use can only be made by empirical testing for related crystal structures.
Reference List
- Price SL, Price LS 2005. Modelling Intermolecular Forces for Organic Crystal Structure Prediction. In Wales DJ, editor. Intermolecular Forces and Clusters I, Berlin, Heidelberg, Germany: Springer-Verlag. p 81-123.
- Price SL 2004. Quantifying intermolecular interactions and their use in computational crystal structure prediction. CrystEngComm 6:344-353.
- Williams DE 2001. Improved intermolecular force field for molecules containing H, C, N, and O atoms, with application to nucleoside and peptide crystals. J Comput Chem 22:1154-1166.
- Day GM, Chisholm J, Shan N, Motherwell WDS, Jones W 2004. Assessment of lattice energy minimization for the prediction of molecular organic crystal structures. Cryst Growth Des 4:1327-1340.
- Day GM, Motherwell WDS, Jones W 2005. Beyond the isotropic atom model in crystal structure prediction of rigid molecules: Atomic multipoles versus point charges. Cryst Growth Des 5:1023-1033.
- Karamertzanis PG 2008. CoCrystal prediction DMAflex. Cryst Growth Des in preparation.
- Williams DE, Cox SR 1984. Nonbonded Potentials For Azahydrocarbons: the Importance of the Coulombic Interaction. Acta Crystallogr , Sect B 40:404-417.
- Coombes DS, Price SL, Willock DJ, Leslie M 1996. Role of Electrostatic Interactions in Determining the Crystal Structures of Polar Organic Molecules. A Distributed Multipole Study. J Phys Chem 100:7352-7360.
- Beyer T, Price SL 2000. Dimer or catemer? Low-energy crystal packings for small carboxylic acids. J Phys Chem B 104:2647-2655.
- Cox SR, Hsu LY, Williams DE 1981. Nonbonded Potential Function Models for Crystalline Oxohydrocarbons. Acta Crystallogr , Sect A 37:293-301.
- Williams DE, Houpt DJ 1986. Fluorine Nonbonded Potential Parameters Derived From Crystalline Perfluorocarbons. Acta Crystallogr , Sect B 42:286-295.
- Hsu LY, Williams DE 1980. Intermolecular Potential-Function Models for Crystalline Perchlorohydrocarbons. Acta Crystallogr , Sect A 36:277-281.
- Hulme AT, Price SL 2007. Towards the prediction of organic hydrate crystal structures. J Chem Theory Comput 3:1597-1608.
- Barnett SA, Johnson A, Florence AJ, Price SL, Tocher DA 2008. A systematic experimental and theoretical study of the crystalline state of six chloronitrobenzenes. Cryst Growth Des 8:24-36.
- Lewis TC, Tocher DA, Price SL 2005. Investigating Unused Hydrogen Bond Acceptors Using Known and Hypothetical Crystal Polymorphism. Cryst Growth Des 5:983-993.
- Lommerse JPM, Motherwell WDS, Ammon HL, Dunitz JD, Gavezzotti A, Hofmann DWM, Leusen FJJ, Mooij WTM, Price SL, Schweizer B, Schmidt MU, van Eijck BP, Verwer P, Williams DE 2000. A test of crystal structure prediction of small organic molecules. Acta Crystallogr, Sect B 56:697-714; Halgren TA 1992. Representation of Vanderwaals (Vdw) Interactions in Molecular Mechanics Force-Fields - Potential Form, Combination Rules, and Vdw Parameters. J Am Chem Soc 114:7827-7843.
- "Table Sheraga" in Supporting Information of Motherwell WDS, Ammon HL, Dunitz JD, Dzyabchenko A, Erk P, Gavezzotti A, Hofmann DWM, Leusen FJJ, Lommerse JPM, Mooij WTM, et al. 2002. Crystal structure prediction of small organic molecules: a second blind test. Acta Crystallogr, Sect B 58:647-661
© UCL Chemistry Department 2022. This page was last updated on 17 August, 2022. If you have any problems with this page please email the WebMaster