|

|
|
| |
Rydberg states are highly excited electronic states in which one electron has been promoted to a high lying electronic state (typically with principal quantum number n > 8). These highly excited states have some remarkable properties, for example, the spatial extent of a Rydberg state scales as the square of the principal quantum number n, so for n = 10, the inner and outer turning points are around 10 nm apart, and for n = 30, they are almost one tenth of a micron apart, which is nearly macroscopic. Another interesting feature is the period of electronic motion. A Rydberg electron with n = 30 will have a classical orbit period of almost 5 ps, which is of the same order of magnitude as the period of molecular rotation in a small molecule, and is around one thousand times slower than a typical molecular vibration. Consequently the Born-Oppenheimer approximation breaks down. Wave packets are spatially localised quantum states possessing both particle-like and wave-like characteristics. They provide an important bridge between quantum mechanics and the classical picture of a particle moving on a classical trajectory. Electronic wave packets created from coherent superpositions of Rydberg states are particularly attractive. Rydberg wave packets in atoms have been the focus of a great deal of interest during the last two decades. Initially, the impetus was on testing the correspondence principle limit of an atom, as invoked by Neils Bohr; although currently, as a result of huge technological advances in ultrafast laser technology, there has been a shift in emphasis from the observation of Rydberg wave packets to their customisation and control. Molecular Rydberg wave packets are even more attractive because of the additional degrees of freedom introduced by the molecular core. These systems exhibit many of the dynamical complications associated with larger molecules of more interest to chemists and biologists, such as the high density of rovibronic states and non-adiabatic coupling, yet they remain highly tractable. In our group we employ frequency-resolved and time-resolved spectroscopy techniques to investigate Rydberg dynamics in atoms and diatomic molecules, both experimentally and theoretically. One of our aims is to take these highly tractable molecular Rydberg systems and design laser pulse sequences or laser pulse shapes to control the non-adiabatic dynamics. Within the last few years we have employed sequences of phase-locked optical pulses to control the orbital angular momentum composition of Rydberg wave packets in Xe and Na, and the rotational angular momentum of Rydberg wave packets in NO. We have also made progress in controlling the autoionisation/predissociation branching ratio in NO. To complement and guide the dynamics experiments we are also interested in the high resolution spectroscopy of highly excited Rydberg states and have recently reported the spectroscopy of Rydberg states of NO in external dc electric fields. As well as using optical pulses to control Rydberg dynamics, we are interested in investigating how DC electric fields can be exploited as control tools. We have recently reported the first observation of the important role that slew rate plays in the field-ionisation of molecular Rydberg species, and demonstrated its potential for controlling the rotational quantum state of the product ions. In collaboration with Professor Christian Jungen (Orsay) we have developed a time-dependent extension of multichannel quantum theory (MQDT) in which time-dependent wave packets are constructed using first-order perturbation theory. We have derived an analytical expression for the complex excitation function for a sequence of excitation pulses and have developed numerical methods to model the complex excitation function for phase and amplitude shaped excitation pulses. Our ultimate goal is to design rational light fields to control the branching ratio of ionisation/dissociation.
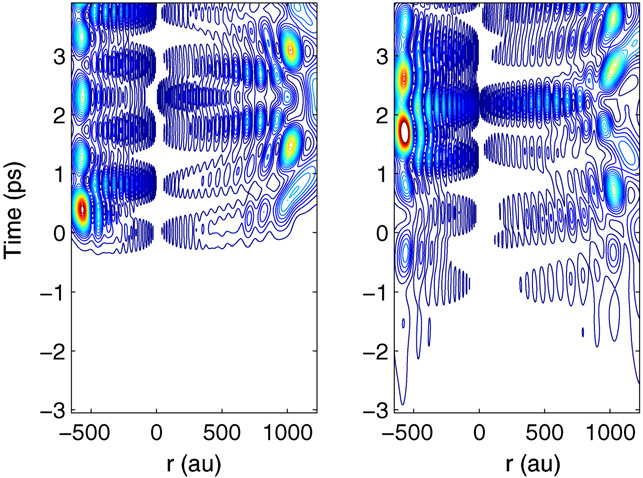
Above: Excitation of a Rydberg electron wave packet in H2 below the lowest ionisation limit. Comparison of the excitation process with the unshaped reference pulse A (left panel) and a phase-shaped pulse B (right panel). The contour plot shows the wave packet probability density as a function of radial distance r (au) and time t (ps). Negative radial distances r correspond to the dynamics in channel j = 2 (ion core rotational quantum number N+ = 2), while positive radial distances correspond to channel j = 1 (N+ = 0). In both channels, the wave packet oscillates between the core (r ≈ 0) and the outer turning point. For pulse A (left panel), the excitation at time t ≈ 0 ps is almost instant and is quickly followed by free evolution of the wave packet. For pulse B (right panel), the excitation process with the shaped pulse begins already at times t < −5 ps, becomes discernible in the contour plot at times t ≈−3 ps and does not evolve freely until for times t > 5 ps. See J. Phys. B. 41 074022 (2008) for details.
Organic
photochemistry Chemists have long dreamt of using laser light to control chemical reactivity; but unfortunately, it is not possible to break a specific bond in a molecule simply by tuning a laser into resonance with a transition to that bond. This is because the energy put into the molecule is redistributed very rapidly on the timescale of molecular vibrations. However, since the advent of femtosecond lasers (1 fs = 0.000 000 000 000 001 s) in the 1980s it has become possible to probe chemical bonds as they are being made and broken and, with further advances in laser technology in the late 1990s, it is now possible to generate complex femtosecond laser pulses that can control these processes. The most common approach to controlling molecular bond breaking has been to employ a learning algorithm and a feedback loop from which the laser optimises its own pulse shape. Such ‘black box’ techniques have been exploited with huge success tocontrol bond dissociation in organometallic complexes. Obviously a great deal of information about the dynamics is concealed within these complex laser pulses but, to date, it has proved difficult to unravel this information. In collaboration with Professor Mike Robb (Imperial), Dr Mike Bearpark (Imperial) and Dr Graham Worth (Birmingham), we are developing the machinery to investigate the dynamics of photochemical reactions in fine detail. Once we have a detailed understanding our aim is to employ shaped light fields to control the dynamics on the excited potential energy surface, and hence control when the molecule returns to the ground state.
Above: Photoelectron imaging vacuum apparatus and uv-femtosecond pulse shaping and characterisation setup. We have recently investigated the ultrafast intramolecular dynamics of electronically and vibrationally excited benzene using time-resolved photoelectron spectroscopy and quantum dynamics simulations. In addition to an ultrafast initial decay we observe an oscillation between two states. The most plausible explanation of the data is that the photophysical pathway involves an ultrafast intersystem crossing from the initially populated singlet state to an optically dark triplet state. Our results challenge the currently accepted view that the initial ultrafast decay from the excited state is dominated by internal conversion through a singlet-singlet conical intersection taking the population directly back to the ground state.
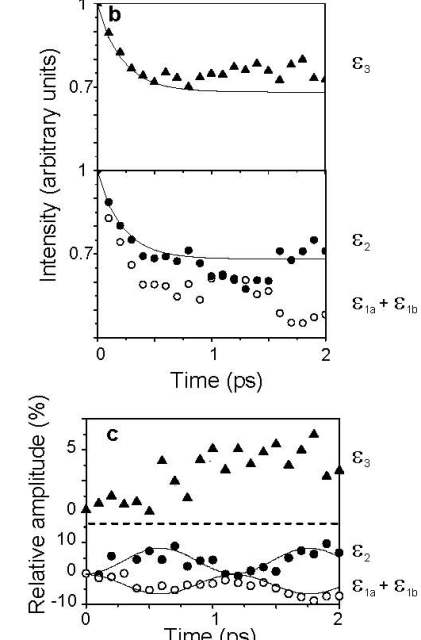
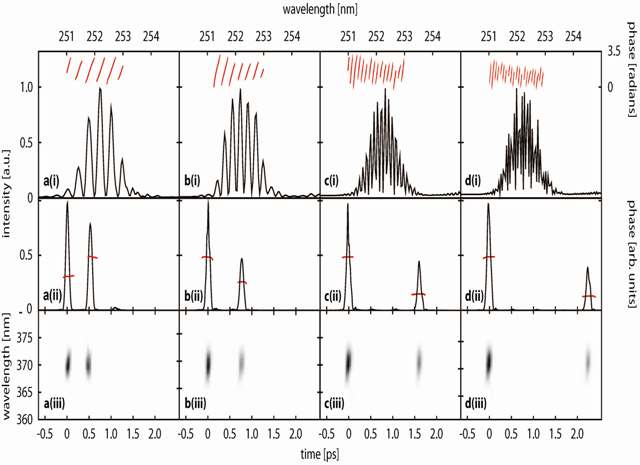
Above: Time-resolved dynamics of benzene during intersystem crossing between S1 and T2 and the subsequent internal conversion to T1. Curves e1a and e1b correspond to photoelectron energies associated with ionisation from S1 and are separated by 0.1 eV, which corresponds to the v1 (a1g) breathing mode in the benzene cation. e2 corresponds to photoelectron energies associated with ionisation from T2 and e3 corresponds to photoelectron energies associated with ionisation from T1. (b) Integrated intensities of photoelectrons as a function of pump-probe delay. The open circles correspond to integration over the range e1a = 1.25-2.35 eV and e1b = 1.13-1.23 eV, and the closed circles correspond to integration over the range e2 = 0.75-0.95 eV. The filled triangles correspond to integration over the range e3 = 0.35-0.45 eV. (c) The same data as (b) but with the exponential decay removed. The solid lines in the lower half of the graph are a least squares sinusoidal fit to the data and have periods 1.2 ps. See Chem. Phys. Lett. 469 43-47 (2009) for details. We have also developed a robust method for producing tuneable, shaped, UV femtosecond laser pulses by shaping and frequency doubling the output of a commercial optical parametric amplifier (OPA). A reflective mode, folded, pulse shaping assembly employing a spatial light modulator (SLM) shapes femtosecond pulses in the visible region of the spectrum. The shaped visible light pulses are frequency doubled to generate phase- and amplitude-shaped, ultrashort light pulses in the deep UV. This approach benefits from a simple experimental setup and the potential for tuning the central frequency of the shaped UV waveform. A number of pulse shapes have been synthesised and characterised using cross-correlation frequency resolved optical gating (XFROG), including pairs of pulses with different central wavelengths.
We are just beginning a new project, in collaboration with Dr Jonathan Underwood, to combine our methods with molecular axis alignment to gain further insight into the dynamics and for developing potential control schemes. This project is funded by the EU Marie-Curie Initial Training Network FASTQUAST. Chromophores in
photobiology Understanding ultrafast processes is of great importance in elucidating protein dynamics. For many biological targets the elementary aspects of these processes are still not well understood. Fluorescent proteins are a good example of this: they are used widely in cell biology, primarily in fluorescence microscopy, yet there are still gaps in our understanding of the fundamental photochemistry and photophysics which can lead to significant pitfalls in applications, ranging from artefacts in measurements to cell death. In contrast to isolated gas-phase systems, the environment of the chromophore in a fluorescent protein plays an essential role in determining the reaction pathway and product distribution. In solution based reactions the solvent molecules move around freely within the solute, whereas in protein based reactions the protein environment provides both a static and dynamical constraint on the motions of the atoms within the chromophore. The protein environment effectively catalyses a photoreaction path that may not be observed in solution at all, but which may be crucial for a specific biological function. In collaboration with Dr Adam McKay and Professor Steve Caddick we are developing a novel experiment to investigate the femtosecond photoinduced dynamics of large proteins, based on a combination of electrospray ionisation mass-spectrometry and time-resolved photoelectron imaging. Our instrument is currently being commissioned and our progress is logged below.
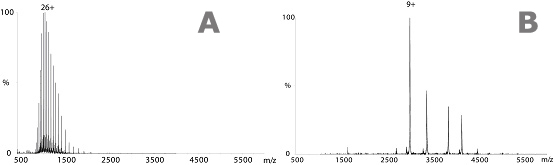
Above: (A) Mass spectrum obtained under denaturing conditions. Protein mass spectrometry experiments used to confirm molecular weights are typically carried out in acidified aqueous/organic solvent solutions. This produces numerous highly charged protein ions and accurate molecular weights, however, the proteins are denatured and all structural information is lost. (B) Mass spectrum of GFP obtained under ‘native-like’ conditions. The use of aqueous buffers such as ammonium acetate allows the protein to be transferred into the gas-phase in a ‘native-like’ state.
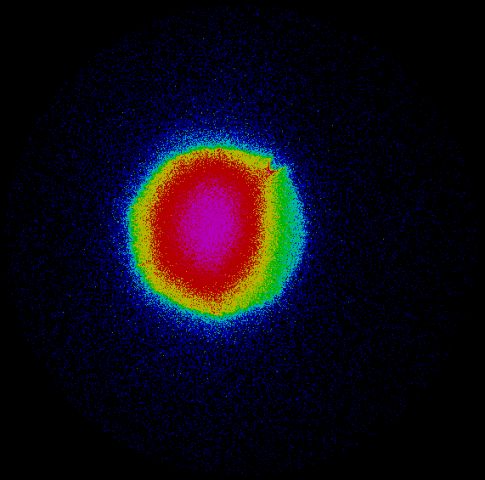
Above: Anions are imaged onto an MCP detector coupled to a phosphore screen and CCD camera (left image). Anions from the pulsed electrospray source are focussed into the source region of a velocity map imaging setup and the two rings in the image on the right arise from photodetachment of the outer electron from I- anions which are being used to calibrate the apparatus. Ultrafast
photochemistry on metal surfaces Surface photochemical processes are an important class of photoreactions, both from a fundamental point of view and also in practical terms. For example, improving our understanding of photon-induced surface processes is a key factor in the design of industrial and environmental catalysts and in the understanding of surface reactions in the atmosphere and in space. Femtosecond laser-induced photochemistry on metal surfaces involves an initial optical excitation of electrons in the metal. This excitation leads to a highly non-equilibrium situation in which the electrons, often referred to as ‘hot’ electrons, have a temperature of several thousand degrees Kelvin. In contrast, the phonon bath within the solid remains relatively cool. On a femtosecond timescale, these hot electrons attach to unoccupied energy levels of the adsorbate. The nuclei are no longer in their equilibrium positions and the adsorbate vibrates, resulting in subsequent desorption or reaction of the adsorbed species. Meanwhile, on a picosecond timescale, the hot electron distribution equilibrates with the lattice and the heated phonon bath can then itself induce desorption and reaction by thermal activation. In collaboration with Dr Wendy Brown we have designed a new experiment to follow the various processes in laser-induced surface reactions in real time, with the ultimate goal of being able to understand and control surface photochemistry. Our experiments employ a novel combination of two-photon correlation (2PC) and time-resolved photoelectron spectroscopy (TRPES). The 2PC technique allows us to determine which reactions are electron and phonon mediated, and to measure the timescale of the reaction processes. In these experiments, the photoreaction yield is measured as a function of the delay time between two, equally intense, laser pulses. Because of the fast coupling between the hot electrons and the phonons, high electronic temperatures result from the combined effect of the two laser pulses, provided that they are separated by a time delay less than the electron-phonon equilibration time of ~1 ps. Hence, a fast response is observed for electron mediated processes. Alternatively, the observation of a slow response implies a phonon mediated process, as the phonon cooling time is ~50 ps. TRPES is used to monitor the real time changes in the electronic structure of the adsorbed reactants and products. For the TRPES experiments a pump pulse, identical to that used in the 2PC experiments, initiates the photochemical reaction. A delayed vacuum ultraviolet (VUV) femtosecond probe pulse will then subsequently ionise the substrate-surface system and the resulting photoelectron spectrum will provide a fingerprint of the valence energy level electronic structure. The probe pulse is a single harmonic of the fundamental generated in a high harmonic generation (HHG) capillary cell, separated from the fundamental and other harmonics using a combination of foils and multilayer reflectors. The kinetic energies of all the photoemitted electrons are measured simultaneously using a commercial hemispherical electron analyser. Our first experiment is to monitor the photo-initiated reaction of CO and NO on a Pd(111) surface. We will then investigate the possibility of coherent control of this reaction using phase shaped infrared femtosecond laser pulses.
Above: Ultrahigh vacuum chamber for time-resolved surface photochemistry. The chamber is equipped for traditional surface science techniques: reflection absorption infra-red spectroscopy, temperature programmed desorption, low energy electron diffraction, auger electron spectroscopy. It also has a hemispherical analyser for time-resolved photoelectron spectroscopy. It will soon be coupled with our source of femtosecond VUV pulses being created by high harmonic generation in a capillary of Ar. Attosecond science All fundamental processes in chemistry, biology and materials science involve the motions of electrons and the nuclei of the constituent atoms. The extremely fast electronic motions have attosecond timescales (1 attosecond = 1 thousandth of a femtosecond). A proper understanding of the way in which electronic motions influence these fundamental processes requires probes on the attosecond timescale. We are interested in exploiting attosecond light sources to investigate organic photochemical reactions. This project is carried out in collaboration with Professor Jon Marangos and Dr John Tisch at Imperial College.
This page last modified 14 November, 2009 by Helen Fielding |